
Per ectasia corneale si intende un gruppo di patologie congenite, a trasmissione ereditaria, caratterizzate dall’alterazione della normale curvatura corneale. Le più frequenti sono: il cheratocono, la degenerazione marginale pellucida, la megalocornea e la microcornea.
Il CHERATOCONO è un’ectasia non infiammatoria caratterizzata da uno sfiancamento ed assottigliamento progressivo del tessuto corneale che fa perdere alla cornea la sua caratteristica forma di calotta sferica per farle assumere la forma di un tronco di cono. Insorge in maniera prevalente nei soggetti di sesso femminile rispetto a quelli di sesso maschile ed è per lo più bilaterale. Non è presente alla nascita, ma si rende manifesto, di solito, verso la pubertà e progredisce sino ad arrestarsi intorno ai quarant'anni, mantenendosi poi stabile per il resto della vita.
L’evoluzione della malattia non è, tuttavia, prevedibile e, a varianti che presentano un esordio più tardivo, si contrappongono forme cronicamente ingravescenti. In genere si presenta bilateralmente sebbene l’evoluzione della malattia nei due occhi sia asincrona. Il cheratocono presenta una frequenza che varia tra lo 0,15% e lo 0,6% della popolazione, ed un’incidenza attorno ad un caso per 2.000 abitanti. L’eziologia non è ancora nota, ma si è concordi sul fatto che, tale malattia, abbia una genesi multifattoriale di cui si ipotizza l’azione di fattori genetici, traumatici, ambientali e biochimici.
La componente genetica è avvalorata da numerose evidenze cliniche e di laboratorio quali: una maggiore incidenza in gemelli monozigoti rispetto a quelli dizigoti ed un’ elevata incidenza della malattia tra i familiari di individui affetti con trasmissione di tipo autosomico dominante a penetranza incompleta. Gli studiosi pongono la loro attenzione sullo studio di diversi geni, in particolare è coinvolto il gene VSX1 localizzato nella regione 20p11-q11, che sintetizza fattori di trascrizione ed è espresso nello strato nucleare della retina e nella cornea, di cui sono state scoperte 4 mutazioni in grado di causare la malattia. Altre mutazione potenzialmente coinvolte nella genesi del cheratocono riguardano il gene della lisil-ossidasi, responsabile del cross-linking del collagene, il gene dalla cell death-inducing DEFA-like effector b, implicato nell'apoptosi, e il gene del gelsolino, associato anche alla distrofia corneale reticolare tipo II.
Tra i fattori traumatici sono da considerare quelli dovuti all’ “eye rubbing ( strofinarsi gli occhi)”, frequente nei soggetti affetti da congiuntivite allergica o congiuntiviti croniche da abuso di lenti a contatto ecc. Tali condizioni causano una morte delle cellule epiteliali che rilasciano interleuchina 1, la quale si lega ai cheratociti causandone la distruzione e quindi l’alterazione della citoarchitettura dello stroma.
In tali soggetti, si è osservato un aumento dell’ incidenza di cheratocono. Tra i fattori ambientali viene considerata l’azione dei raggi UV i quali, a contatto con il tessuto corneale, determinano il rilascio di radicali liberi. Questi ultimi determinano una perossidazione lipidica danneggiando le attività enzimatiche cellulari, la sintesi del DNA e dell’ RNA con conseguente danno e morte cellulare. L’ azione tossica di tali radicali liberi è contrastata dalla presenza di enzimi antiossidanti (superossido dismutasi, catalasi, ALDH3 ecc.) che risultano carenti nella cornea affetta da cheratocono. Gli studi dei fattori biochimici, infine, hanno messo in evidenza, nei soggetti affetti, l’alterazione del collagene corneale. Nel cheratocono la composizione del collagene è uguale a quella della cornea sana ma sono evidenti delle alterazioni a carico delle lamelle che appaiono fuse e degenerate. Dall’analisi biochimica di queste lamelle si è visto che esse sono costituite da catene di cheratansolfato più corte e con peso molecolare inferiore rispetto a quelle di cornee sane. Secondo alcuni autori l’alterazione primaria potrebbe ricercarsi in un aumento del rapporto tra glicoproteine e mucopolisaccaridi nella cornea colpita: tale condizione induce una desincronizzazione della velocità relativa di biosintesi del collagene e delle glicoproteine strutturali da parte dei cheratociti.
Si è osservata una forte diminuzione del tasso di glucosio 6-P deidrogenasi nell’epitelio di pazienti affetti da cheratocono; ciò si correla ad una anomala ossidazione diretta del glucosio attraverso lo shunt del pentoso-fosfato. Una diminuzione del pentoso disponibile per la sintesi dell’RNA potrebbe ostacolare la formazione del collagene che rappresenta il costituente strutturale essenziale dello stroma, determinandone così un progressivo assottigliamento.
Teorie più recenti focalizzano l’attenzione sul binomio proteasi-antiproteasi. Secondo tali studi la patologia sarebbe il frutto di un’alterazione della normale condizione di equilibrio esistente tra strutture enzimatiche presenti a livello dei cheratociti ed i propri inibitori. Tale squilibrio potrebbe riconoscere come fattore causale sia un aumento del tasso enzimatico, sia una riduzione del tasso delle antiproteasi. In riferimento alla prima ipotesi è stato possibile apprezzare in diversi studi, l’incremento del tasso di enzimi lisosomiali, in particolare delle catepsine B e G responsabili di lesioni stromali e di lesioni a livello della membrana di Bowman. A ciò si aggiungono i numerosi approfondimenti che testimoniano un notevole incremento nelle cornee affette da cheratocono, del tasso di metalloproteinasi della matrice extracellulare; si tratta sostanzialmente di proteasi a diversa specificità, in particolare di collagenasi e quindi di enzimi deputati al catabolismo delle fibre collagene che costituiscono il tessuto corneale. Per contro recentissimi studi riconducono l’esaltata attività enzimatica non ad un primitivo patologico incremento delle metalloproteinasi bensì ad una drastica riduzione dei corrispettivi sistemi enzimatici di inibizione. Tenendo conto di tutte queste osservazioni si può, dunque, ipotizzare che il cheratocono sia causato dall’alterazione di uno o più geni che con un meccanismo non ancora chiarito, e con la partecipazione di fenomeni ambientali e traumatici, determinerebbero uno squilibrio fra la produzione e la degradazione delle fibre collagene dello stroma con conseguente riduzione dello spessore corneale ed alterazione della capacità di resistenza tensionale; queste caratteristiche avrebbero l'effetto di causare la deformazione corneale nel corso degli anni.
Dal punto di vista anatomo-patologico il cheratocono si caratterizza per una serie di alterazioni che coinvolgono in toto la cornea; in particolare, a livello epiteliale sarà possibile apprezzare un progressivo accumulo di materiale ferroso con disorganizzazione delle cellule dello strato basale che si rigonfiano e tendono a scomparire, mentre la membrana basale va incontro a frammentazioni multiple; la membrana di Bowman presenterà rigonfiamento e degenerazione fibrillare con soluzioni di continuo multiple riempite da tessuto connettivo e fibroblasti (ingrowth epiteliale); a livello stromale sarà visibile un assottigliamento e riduzione delle lamelle con aree di degenerazione fibrillare, distruzione e sostituzione con tessuto connettivo intercalate ad accumuli di sostanza amiloide (frammentazione fibrillare). Peculiari sono le anomalie apprezzabili a livello endoteliale sia con cellule che tendono ad appiattirsi e con nuclei che tendono a distanziarsi tra loro (polimegatismo), sia con l’assottigliamento e la rottura della membrana di Descemet (retrazione elastica).
Dal punto di vista clinico si osserva un progressivo sfiancamento corneale che genera un astigmatismo irregolare ed un’ alterazione del tessuto corneale in toto. La sintomatologia, quindi, sarà legata all’entità dell’astigmatismo e sarà dominata da una progressiva, ed inesorabile, diminuzione del visus. Mentre i segni obiettivi della malattia, legati allo stadio di alterazione strutturale della cornea, sono caratterizzati dalla presenza di strie stromali conseguenza della frammentazione fibrillare (strie di Vogt), opacità e nubecole all’apice conseguenza della deposizione di materiale ialino e cheratociti degenerati, fino ad arrivare all’edema diffuso conseguente alla sofferenza delle cellule endoteliali ed alla retrazione della membrana di Deschemet.
Le caratteristiche evolutive della malattia necessitano la codificazione di una stadiazione al fine sia di inquadrare al meglio la malattia, sia di programmare il miglior atto terapeutico. Esistono numerose stadiazioni tra le quali la più recente è quella di Krumeich:
· Stadio I: miopia e astigmatismo indotto < 5D, K reading < 48 D, assenza di cicatrici corneali, pachimetria > 500 micron;
· Stadio II: miopia e/o astigmatismo indotto > 5 < 8 D, K reading < 53 D, assenza di cicatrici corneali, pachimetria > 400 micron;
· Stadio III: miopia e/o astigmatismo indotto > 8 < 10 D, K reading > 53 D, assenza di cicatrici corneali, pachimetria 200-400 micron;
· Stadio IV: refrazione non misurabile, K reading > 55 D, presenza di cicatrici corneali centrali e spessore corneale < 200 micron.
(Fonte: sito SOI - www.soiweb.com)
DIAGNOSI
Le tecniche diagnostiche si avvalgono di:
· oftalmoscopia diretta: il riflesso retinico risulta più chiaro nella zona periferica e più scuro dove è posizionato il cono
· oftalmometro di Javal: con cui valutare l'angolo di Amsler
· retinoscopio a striscia: evidenzia il riflesso a forbice
· lampada a fessura o biomicroscopio: permette di ricercare i seguenti reperti: l'anello di Fleischer che è patognomonico (presente nel 50% dei KC): è un anello giallo-verdastro dovuto all’accumulo di emosiderina nelle cellule epiteliali basali
· opacità subepiteliali (che si formano da rotture della membrana di Bowman)
· assottigliamento stromale: L’assottigliamento sarà maggiore a livello dell’apice del cono
· strie di Vogt (tardive): strie stromali che scompaiono alla digitopressione, hanno aspetto verticale
· segno di Munson (avanzato): deformazione della rima palpebrale inferiore da parte del cono e si visualizza facendo guardare verso il basso il soggetto
· pachimetria
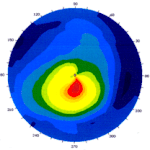
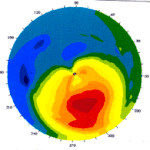
· topografia corneale (gold standard): studia le immagini riflesse dalla cornea quando le si antepone una serie di anelli luminosi concentrici; L'elaborazione dei dati consente la visualizzazione di una mappa (la parte in rosso corrisponde alla regione sfiancata mentre quella verde-blu le zone piatte) e di una simulazione della qualità della visione consentita da tale cornea. Permette di vedere l’aumento di curvatura negli stadi iniziali ed avanzati e permette la diagnosi precoce (NB inizialmente il cheratocono non si vede alla lampada a fessura)
· aberrometria: sintomi tipici sono l'aberrazione della coma verticale e l'aberrazione sferica che porta alla percezione di immagini-fantasma, e di aloni intorno alle sorgenti luminose;
· microscopia confocale (Confoscan di Nidek): una sorta di esame istologico in vivo delle lesioni tipiche del KC
· immagini della cornea e del segmento anteriore ottenute l'ecografia ad alta frequenza
TERAPIA
Nelle forme lievi si utilizzano occhiali o lenti a contatto rigide o semirigide per correggere il difetto visivo.
Un trattamento chirurgico relativamente recente è rappresentato dagli inserti intracorneali (ICRS). La tecnica, che migliora sensibilmente l'acuità visiva dei pazienti (visus), consiste nell’impianto di microscopici inserti in materiale sintetico trasparente appena sotto la superficie dell’occhio, alla periferia della cornea. Un'ulteriore opzione è la Mini Cheratotomia Radiale Asimmetrica (M.A.R.K.), tecnica chirurgica incisionale che provoca una cicatrizzazione controllata della cornea, la quale cambia forma e spessore secondo il bisogno dell'occhio affetto da cheratocono. Questa tecnica può essere combinata con il cross-linking al fine di rinforzare ulteriormente la cornea.
Le forme più gravi tuttavia, che sono progressive e conducono a uno sfiancamento e assottigliamento della cornea, o il cheratoglobo, necessitano invece di un intervento chirurgico radicale come il trapianto di cornea.
Il cross-linking corneale
Nel 1997 viene inventato, presso l'università di Dresda in Germania, il cross-linking corneale. In Italia viene applicato dal 2005, oggi viene utilizzato in molti Paesi del mondo.
Nel 2005 il Policlinico di Siena ha iniziato in Italia una investigazione sugli effetti che tale tecnica ha sul collagene corneale. Per compiere questa ricerca si è utilizzato uno strumento noto come microscopio confocale.
La tecnica, avviata in Germania nel 1997 ma diffusa solo negli ultimi anni, consiste nell'instillare delle gocce di vitamina B2 (riboflavina) sulla cornea con epitelio rimosso e, contemporaneamente, esporre la cornea a una luce ultravioletta. La reazione chimica dei raggi UV-A che stimolano la riboflavina comporta un rafforzamento dei legami nel collagene corneale con un conseguente indurimento della cornea. Gli studi hanno dimostrato che si riesce a bloccare l'evoluzione della malattia e, in molti casi, si verifica una diminuzione della curvatura della cornea (2 diottrie in media).
La tecnica è esente da rischi se viene rispettato il protocollo ideato, e ampiamente sperimentato, a Dresda.
Alcuni oculisti italiani hanno cominciato ad apportare delle modifiche a tale protocollo ma l'efficacia di queste modifiche è stata fortemente messa in dubbio nel corso del congresso tenutosi a Dresda nel dicembre 2008, in occasione del decennale dell'applicazione del cross-linking.
L'utilizzo di colliri diversi dall'originale e l'impiego di macchinari diversi da quello ideato a Dresda potrebbe portare a una minore efficacia del trattamento stesso.
Nel 2006 è partito uno studio multicentrico che ha coinvolto, oltre all'Università di Siena, molti altri centri oculistici italiani.
Dal 1º gennaio 2007, tale terapia è stata riconosciuta a livello sia nazionale che sovranazionale (Unione europea) come cura ufficiale. Nel corso del 2007, grazie agli eccezionali risultati riconosciuti internazionalmente, si sono moltiplicati i centri che praticano il cross-linking. Tuttavia non dappertutto viene effettuato in regime di completa gratuità, ma solo in alcune regioni dove viene rimborsato dal Servizio Sanitario Regionale. In Toscana il costo del trattamento è interamente sostenuto dal Sistema Sanitario Regionale (si paga solo il ticket). Nel Lazio il trattamento viene eseguito a totale carico del Servizio Sanitario Nazionale presso l'Ospedale oftalmico regionale di Roma. È da tenere, tuttavia, presente che, per poter effettuare tale tecnica, ci sono dei limiti (ad esempio limiti morfologici: il cheratocono non deve essere troppo evoluto e lo spessore della cornea deve essere soddisfacente).
Il cross-linking corneale può, in alcuni casi, essere combinato con altre procedure di chirurgia conservativa del cheratocono. Il Medico Oculista consiglia quale paziente è idoneo all'intervento e prima dello stesso esegue un completo esame dl bulbo oculare oltre agli approfondimenti diagnostici necessari. Come ogni altro intervento chirurgico, anche il cross.linking non è scevro da possibili complicanze (es. "haze" corneale).
Cheratoplastica lamellare
Introdotta a partire dal 1998, consente di sostituire non l'intera cornea, ma solo la parte più esterna, quella affetta dalla malattia.
La DEGENERAZIONE MARGINALE PELLUCIDA rappresenta un’ectasia caratterizzata da uno sfiancamento ed assottigliamento dei quadranti inferiori della cornea. Si tratta di una patologia bilaterale anche se spesso asimmetrica e risulta asintomatica finchè non insorge un disturbo del visus legato all’elevato astigmatismo irregolare. La topografia corneale, invece, mostra una tipica forma “a farfalla”. La sintomatologia e la presentazione clinica sono sovrapponibili a quelle del cheratocono salvo che per il fatto che, a differenza del primo, lo sfiancamento è prevalentemente inferiore. La diagnosi differenziale avviene mediante la topografia che presenterà il classico aspetto a farfalla. Come per il cheratocono, non esiste una terapia medica. Il trattamento ottico prevede l’impiego sia di occhiali che di lenti a contatto rigide. La terapia chirurgica è affidata al trapianto di cornea che, però, non ottiene gli stessi risultati come nel caso del cheratocono.
La MEGALOCORNEA o CHERATOMEGALIA è una patologia bilaterale trasmessa come carattere autosomico recessivo legato al cromosoma X e colpisce soggetti di sesso maschile. Si presenta con un segmento anteriore dell’occhio di dimensioni maggiori con maggiore grandezza di cornea, cristallino, zonula e corpo ciliare. La cornea mantiene sempre la sua normale trasparenza ed è presente un astigmatismo secondo regola; l’iride mostra aree di subatrofia dello stroma e può presentare piccoli movimenti (iridodonesi). È importante effettuare la diagnosi differenziale con il glaucoma congenito malformativo o buftalmo; tuttavia una pressione intraoculare normale, l’assenza di malformazioni dell’angolo camerulare, di alterazioni della trasparenza della cornea e di alterazioni della papilla ottica depongono per una condizione di megalocornea.
La MICROCORNEA è invece legata ad un arresto dello sviluppo dell’organogenesi dopo il 5°mese di vita. È caratterizzata da una normale grandezza dell’occhio con cornea di diametro inferiore agli 11 mm. Si associa ad astigmatismo regolare e ad un’ipermetropia media correggibile con lenti sferiche positive o talvolta ad emmetropia. Tale condizione non va però confusa con la cornea piccola del microftalmo in cui si ha riduzione di tutti i diametri oculari.
Per ectasia corneale si intende un gruppo di patologie congenite, a trasmissione ereditaria, caratterizzate dall’alterazione della normale curvatura corneale. Le più frequenti sono: il cheratocono, la degenerazione marginale pellucida, la megalocornea e la microcornea.
Il CHERATOCONO è un’ectasia non infiammatoria caratterizzata da uno sfiancamento ed assottigliamento progressivo del tessuto corneale che fa perdere alla cornea la sua caratteristica forma di calotta sferica per farle assumere la forma di un tronco di cono. Insorge in maniera prevalente nei soggetti di sesso femminile rispetto a quelli di sesso maschile ed è per lo più bilaterale. Non è presente alla nascita, ma si rende manifesto, di solito, verso la pubertà e progredisce sino ad arrestarsi intorno ai quarant'anni, mantenendosi poi stabile per il resto della vita.
L’evoluzione della malattia non è, tuttavia, prevedibile e, a varianti che presentano un esordio più tardivo, si contrappongono forme cronicamente ingravescenti. In genere si presenta bilateralmente sebbene l’evoluzione della malattia nei due occhi sia asincrona. Il cheratocono presenta una frequenza che varia tra lo 0,15% e lo 0,6% della popolazione, ed un’incidenza attorno ad un caso per 2.000 abitanti. L’eziologia non è ancora nota, ma si è concordi sul fatto che, tale malattia, abbia una genesi multifattoriale di cui si ipotizza l’azione di fattori genetici, traumatici, ambientali e biochimici.
La componente genetica è avvalorata da numerose evidenze cliniche e di laboratorio quali: una maggiore incidenza in gemelli monozigoti rispetto a quelli dizigoti ed un’ elevata incidenza della malattia tra i familiari di individui affetti con trasmissione di tipo autosomico dominante a penetranza incompleta. Gli studiosi pongono la loro attenzione sullo studio di diversi geni, in particolare è coinvolto il gene VSX1 localizzato nella regione 20p11-q11, che sintetizza fattori di trascrizione ed è espresso nello strato nucleare della retina e nella cornea, di cui sono state scoperte 4 mutazioni in grado di causare la malattia. Altre mutazione potenzialmente coinvolte nella genesi del cheratocono riguardano il gene della lisil-ossidasi, responsabile del cross-linking del collagene, il gene dalla cell death-inducing DEFA-like effector b, implicato nell'apoptosi, e il gene del gelsolino, associato anche alla distrofia corneale reticolare tipo II.
Tra i fattori traumatici sono da considerare quelli dovuti all’ “eye rubbing ( strofinarsi gli occhi)”, frequente nei soggetti affetti da congiuntivite allergica o congiuntiviti croniche da abuso di lenti a contatto ecc. Tali condizioni causano una morte delle cellule epiteliali che rilasciano interleuchina 1, la quale si lega ai cheratociti causandone la distruzione e quindi l’alterazione della citoarchitettura dello stroma.
In tali soggetti, si è osservato un aumento dell’ incidenza di cheratocono. Tra i fattori ambientali viene considerata l’azione dei raggi UV i quali, a contatto con il tessuto corneale, determinano il rilascio di radicali liberi. Questi ultimi determinano una perossidazione lipidica danneggiando le attività enzimatiche cellulari, la sintesi del DNA e dell’ RNA con conseguente danno e morte cellulare. L’ azione tossica di tali radicali liberi è contrastata dalla presenza di enzimi antiossidanti (superossido dismutasi, catalasi, ALDH3 ecc.) che risultano carenti nella cornea affetta da cheratocono. Gli studi dei fattori biochimici, infine, hanno messo in evidenza, nei soggetti affetti, l’alterazione del collagene corneale. Nel cheratocono la composizione del collagene è uguale a quella della cornea sana ma sono evidenti delle alterazioni a carico delle lamelle che appaiono fuse e degenerate. Dall’analisi biochimica di queste lamelle si è visto che esse sono costituite da catene di cheratansolfato più corte e con peso molecolare inferiore rispetto a quelle di cornee sane. Secondo alcuni autori l’alterazione primaria potrebbe ricercarsi in un aumento del rapporto tra glicoproteine e mucopolisaccaridi nella cornea colpita: tale condizione induce una desincronizzazione della velocità relativa di biosintesi del collagene e delle glicoproteine strutturali da parte dei cheratociti.
Si è osservata una forte diminuzione del tasso di glucosio 6-P deidrogenasi nell’epitelio di pazienti affetti da cheratocono; ciò si correla ad una anomala ossidazione diretta del glucosio attraverso lo shunt del pentoso-fosfato. Una diminuzione del pentoso disponibile per la sintesi dell’RNA potrebbe ostacolare la formazione del collagene che rappresenta il costituente strutturale essenziale dello stroma, determinandone così un progressivo assottigliamento.
Teorie più recenti focalizzano l’attenzione sul binomio proteasi-antiproteasi. Secondo tali studi la patologia sarebbe il frutto di un’alterazione della normale condizione di equilibrio esistente tra strutture enzimatiche presenti a livello dei cheratociti ed i propri inibitori. Tale squilibrio potrebbe riconoscere come fattore causale sia un aumento del tasso enzimatico, sia una riduzione del tasso delle antiproteasi. In riferimento alla prima ipotesi è stato possibile apprezzare in diversi studi, l’incremento del tasso di enzimi lisosomiali, in particolare delle catepsine B e G responsabili di lesioni stromali e di lesioni a livello della membrana di Bowman. A ciò si aggiungono i numerosi approfondimenti che testimoniano un notevole incremento nelle cornee affette da cheratocono, del tasso di metalloproteinasi della matrice extracellulare; si tratta sostanzialmente di proteasi a diversa specificità, in particolare di collagenasi e quindi di enzimi deputati al catabolismo delle fibre collagene che costituiscono il tessuto corneale. Per contro recentissimi studi riconducono l’esaltata attività enzimatica non ad un primitivo patologico incremento delle metalloproteinasi bensì ad una drastica riduzione dei corrispettivi sistemi enzimatici di inibizione. Tenendo conto di tutte queste osservazioni si può, dunque, ipotizzare che il cheratocono sia causato dall’alterazione di uno o più geni che con un meccanismo non ancora chiarito, e con la partecipazione di fenomeni ambientali e traumatici, determinerebbero uno squilibrio fra la produzione e la degradazione delle fibre collagene dello stroma con conseguente riduzione dello spessore corneale ed alterazione della capacità di resistenza tensionale; queste caratteristiche avrebbero l'effetto di causare la deformazione corneale nel corso degli anni.
Dal punto di vista anatomo-patologico il cheratocono si caratterizza per una serie di alterazioni che coinvolgono in toto la cornea; in particolare, a livello epiteliale sarà possibile apprezzare un progressivo accumulo di materiale ferroso con disorganizzazione delle cellule dello strato basale che si rigonfiano e tendono a scomparire, mentre la membrana basale va incontro a frammentazioni multiple; la membrana di Bowman presenterà rigonfiamento e degenerazione fibrillare con soluzioni di continuo multiple riempite da tessuto connettivo e fibroblasti (ingrowth epiteliale); a livello stromale sarà visibile un assottigliamento e riduzione delle lamelle con aree di degenerazione fibrillare, distruzione e sostituzione con tessuto connettivo intercalate ad accumuli di sostanza amiloide (frammentazione fibrillare). Peculiari sono le anomalie apprezzabili a livello endoteliale sia con cellule che tendono ad appiattirsi e con nuclei che tendono a distanziarsi tra loro (polimegatismo), sia con l’assottigliamento e la rottura della membrana di Descemet (retrazione elastica).
Dal punto di vista clinico si osserva un progressivo sfiancamento corneale che genera un astigmatismo irregolare ed un’ alterazione del tessuto corneale in toto. La sintomatologia, quindi, sarà legata all’entità dell’astigmatismo e sarà dominata da una progressiva, ed inesorabile, diminuzione del visus. Mentre i segni obiettivi della malattia, legati allo stadio di alterazione strutturale della cornea, sono caratterizzati dalla presenza di strie stromali conseguenza della frammentazione fibrillare (strie di Vogt), opacità e nubecole all’apice conseguenza della deposizione di materiale ialino e cheratociti degenerati, fino ad arrivare all’edema diffuso conseguente alla sofferenza delle cellule endoteliali ed alla retrazione della membrana di Deschemet.
Le caratteristiche evolutive della malattia necessitano la codificazione di una stadiazione al fine sia di inquadrare al meglio la malattia, sia di programmare il miglior atto terapeutico. Esistono numerose stadiazioni tra le quali la più recente è quella di Krumeich:
· Stadio I: miopia e astigmatismo indotto < 5D, K reading < 48 D, assenza di cicatrici corneali, pachimetria > 500 micron;
· Stadio II: miopia e/o astigmatismo indotto > 5 < 8 D, K reading < 53 D, assenza di cicatrici corneali, pachimetria > 400 micron;
· Stadio III: miopia e/o astigmatismo indotto > 8 < 10 D, K reading > 53 D, assenza di cicatrici corneali, pachimetria 200-400 micron;
· Stadio IV: refrazione non misurabile, K reading > 55 D, presenza di cicatrici corneali centrali e spessore corneale < 200 micron.
(Fonte: sito SOI - www.soiweb.com)
DIAGNOSI
Le tecniche diagnostiche si avvalgono di:
· oftalmoscopia diretta: il riflesso retinico risulta più chiaro nella zona periferica e più scuro dove è posizionato il cono
· oftalmometro di Javal: con cui valutare l'angolo di Amsler
· retinoscopio a striscia: evidenzia il riflesso a forbice
· lampada a fessura o biomicroscopio: permette di ricercare i seguenti reperti: l'anello di Fleischer che è patognomonico (presente nel 50% dei KC): è un anello giallo-verdastro dovuto all’accumulo di emosiderina nelle cellule epiteliali basali
· opacità subepiteliali (che si formano da rotture della membrana di Bowman)
· assottigliamento stromale: L’assottigliamento sarà maggiore a livello dell’apice del cono
· strie di Vogt (tardive): strie stromali che scompaiono alla digitopressione, hanno aspetto verticale
· segno di Munson (avanzato): deformazione della rima palpebrale inferiore da parte del cono e si visualizza facendo guardare verso il basso il soggetto
· pachimetria
· topografia corneale (gold standard): studia le immagini riflesse dalla cornea quando le si antepone una serie di anelli luminosi concentrici; L'elaborazione dei dati consente la visualizzazione di una mappa (la parte in rosso corrisponde alla regione sfiancata mentre quella verde-blu le zone piatte) e di una simulazione della qualità della visione consentita da tale cornea. Permette di vedere l’aumento di curvatura negli stadi iniziali ed avanzati e permette la diagnosi precoce (NB inizialmente il cheratocono non si vede alla lampada a fessura)
· aberrometria: sintomi tipici sono l'aberrazione della coma verticale e l'aberrazione sferica che porta alla percezione di immagini-fantasma, e di aloni intorno alle sorgenti luminose;
· microscopia confocale (Confoscan di Nidek): una sorta di esame istologico in vivo delle lesioni tipiche del KC
· immagini della cornea e del segmento anteriore ottenute l'ecografia ad alta frequenza
TERAPIA
Nelle forme lievi si utilizzano occhiali o lenti a contatto rigide o semirigide per correggere il difetto visivo.
Un trattamento chirurgico relativamente recente è rappresentato dagli inserti intracorneali (ICRS). La tecnica, che migliora sensibilmente l'acuità visiva dei pazienti (visus), consiste nell’impianto di microscopici inserti in materiale sintetico trasparente appena sotto la superficie dell’occhio, alla periferia della cornea. Un'ulteriore opzione è la Mini Cheratotomia Radiale Asimmetrica (M.A.R.K.), tecnica chirurgica incisionale che provoca una cicatrizzazione controllata della cornea, la quale cambia forma e spessore secondo il bisogno dell'occhio affetto da cheratocono. Questa tecnica può essere combinata con il cross-linking al fine di rinforzare ulteriormente la cornea.
Le forme più gravi tuttavia, che sono progressive e conducono a uno sfiancamento e assottigliamento della cornea, o il cheratoglobo, necessitano invece di un intervento chirurgico radicale come il trapianto di cornea.
Il cross-linking corneale
Nel 1997 viene inventato, presso l'università di Dresda in Germania, il cross-linking corneale. In Italia viene applicato dal 2005, oggi viene utilizzato in molti Paesi del mondo.
Nel 2005 il Policlinico di Siena ha iniziato in Italia una investigazione sugli effetti che tale tecnica ha sul collagene corneale. Per compiere questa ricerca si è utilizzato uno strumento noto come microscopio confocale.
La tecnica, avviata in Germania nel 1997 ma diffusa solo negli ultimi anni, consiste nell'instillare delle gocce di vitamina B2 (riboflavina) sulla cornea con epitelio rimosso e, contemporaneamente, esporre la cornea a una luce ultravioletta. La reazione chimica dei raggi UV-A che stimolano la riboflavina comporta un rafforzamento dei legami nel collagene corneale con un conseguente indurimento della cornea. Gli studi hanno dimostrato che si riesce a bloccare l'evoluzione della malattia e, in molti casi, si verifica una diminuzione della curvatura della cornea (2 diottrie in media).
La tecnica è esente da rischi se viene rispettato il protocollo ideato, e ampiamente sperimentato, a Dresda.
Alcuni oculisti italiani hanno cominciato ad apportare delle modifiche a tale protocollo ma l'efficacia di queste modifiche è stata fortemente messa in dubbio nel corso del congresso tenutosi a Dresda nel dicembre 2008, in occasione del decennale dell'applicazione del cross-linking.
L'utilizzo di colliri diversi dall'originale e l'impiego di macchinari diversi da quello ideato a Dresda potrebbe portare a una minore efficacia del trattamento stesso.
Nel 2006 è partito uno studio multicentrico che ha coinvolto, oltre all'Università di Siena, molti altri centri oculistici italiani.
Dal 1º gennaio 2007, tale terapia è stata riconosciuta a livello sia nazionale che sovranazionale (Unione europea) come cura ufficiale. Nel corso del 2007, grazie agli eccezionali risultati riconosciuti internazionalmente, si sono moltiplicati i centri che praticano il cross-linking. Tuttavia non dappertutto viene effettuato in regime di completa gratuità, ma solo in alcune regioni dove viene rimborsato dal Servizio Sanitario Regionale. In Toscana il costo del trattamento è interamente sostenuto dal Sistema Sanitario Regionale (si paga solo il ticket). Nel Lazio il trattamento viene eseguito a totale carico del Servizio Sanitario Nazionale presso l'Ospedale oftalmico regionale di Roma. È da tenere, tuttavia, presente che, per poter effettuare tale tecnica, ci sono dei limiti (ad esempio limiti morfologici: il cheratocono non deve essere troppo evoluto e lo spessore della cornea deve essere soddisfacente).
Il cross-linking corneale può, in alcuni casi, essere combinato con altre procedure di chirurgia conservativa del cheratocono. Il Medico Oculista consiglia quale paziente è idoneo all'intervento e prima dello stesso esegue un completo esame dl bulbo oculare oltre agli approfondimenti diagnostici necessari. Come ogni altro intervento chirurgico, anche il cross.linking non è scevro da possibili complicanze (es. "haze" corneale).
Cheratoplastica lamellare
Introdotta a partire dal 1998, consente di sostituire non l'intera cornea, ma solo la parte più esterna, quella affetta dalla malattia.
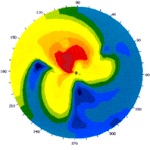
La DEGENERAZIONE MARGINALE PELLUCIDA rappresenta un’ectasia caratterizzata da uno sfiancamento ed assottigliamento dei quadranti inferiori della cornea. Si tratta di una patologia bilaterale anche se spesso asimmetrica e risulta asintomatica finchè non insorge un disturbo del visus legato all’elevato astigmatismo irregolare. La topografia corneale, invece, mostra una tipica forma “a farfalla”. La sintomatologia e la presentazione clinica sono sovrapponibili a quelle del cheratocono salvo che per il fatto che, a differenza del primo, lo sfiancamento è prevalentemente inferiore. La diagnosi differenziale avviene mediante la topografia che presenterà il classico aspetto a farfalla. Come per il cheratocono, non esiste una terapia medica. Il trattamento ottico prevede l’impiego sia di occhiali che di lenti a contatto rigide. La terapia chirurgica è affidata al trapianto di cornea che, però, non ottiene gli stessi risultati come nel caso del cheratocono.
La MEGALOCORNEA o CHERATOMEGALIA è una patologia bilaterale trasmessa come carattere autosomico recessivo legato al cromosoma X e colpisce soggetti di sesso maschile. Si presenta con un segmento anteriore dell’occhio di dimensioni maggiori con maggiore grandezza di cornea, cristallino, zonula e corpo ciliare. La cornea mantiene sempre la sua normale trasparenza ed è presente un astigmatismo secondo regola; l’iride mostra aree di subatrofia dello stroma e può presentare piccoli movimenti (iridodonesi). È importante effettuare la diagnosi differenziale con il glaucoma congenito malformativo o buftalmo; tuttavia una pressione intraoculare normale, l’assenza di malformazioni dell’angolo camerulare, di alterazioni della trasparenza della cornea e di alterazioni della papilla ottica depongono per una condizione di megalocornea.
La MICROCORNEA è invece legata ad un arresto dello sviluppo dell’organogenesi dopo il 5°mese di vita. È caratterizzata da una normale grandezza dell’occhio con cornea di diametro inferiore agli 11 mm. Si associa ad astigmatismo regolare e ad un’ipermetropia media correggibile con lenti sferiche positive o talvolta ad emmetropia. Tale condizione non va però confusa con la cornea piccola del microftalmo in cui si ha riduzione di tutti i diametri oculari.


















Per APPUNTAMENTI
si prega di
contattare il
3343695166
dal lunedì al venerdì
dalle 9,00 alle 13,00
e
dalle 14,30 alle 18,30
Oppure inviare un SMS o un messaggio WhatsApp con la Vostra richiesta e sarete ricontattati